
随着单细胞测序技术的快速发展,多个全球性的、大规模的细胞图谱项目相继启动,例如人类细胞图谱计划等等。然而,把来自于不同组织、不同发育阶段或者只有很少重叠细胞类型的单细胞转录组数据集进行准确有效的整合仍然具有非常大的挑战性。因为当前整合单细胞数据的方法需要校正批次效应从而达到整合分析的目的,而上述所涉及的数据集的批次信息不清晰,利用这些方法常常会导致校正过度或者校正不足等问题,从而影响研究结论的准确性和可靠性。

为了解决上述问题,2021年9月21日,北京未来基因诊断高精尖创新中心、北京大学生物医学前沿创新中心汤富酬课题组与北京大学数学学院李铁军课题组合作在Briefings in Bioinformatics上在线发表了题为Integrating single-cell datasets with ambiguous batch information by incorporating molecular network features的研究论文。该研究引入了一个名为SCORE(Single-Cell mOleculaR nEtwork)的统一分析框架,该方法可以不依赖批次信息整合不同单细胞数据集(https://github.com/wycwycpku/RSCORE)。
该研究假设各个基因在细胞中不是孤立地执行功能,而是倾向于多个不同的基因形成复杂的分子网络(例如不同基因的蛋白质产物相互结合形成蛋白质复合物)、相互作用一起来决定细胞或生物体的生理表型。而且,在生物体的发育和分化过程中,从一种细胞状态到另一种细胞状态的转变常常伴随着对前一个细胞状态关键分子网络的主动抑制以及对下一个细胞状态分子网络的协同激活(图一)。基于以上的科学假设,SCORE精准模拟了单细胞数据集的分子网络动态变化,成功地整合了人类胚胎5个不同器官的单细胞转录组数据集和人类成年个体的15 个主要器官的单细胞转录组数据集。除此之外,该研究还验证了SCORE的准确性、稳健性和可扩展性。综上所述,SCORE 可以帮助整合和分析从各种来源获得的单细胞转录组、染色质状态组等各种组学的数据集(例如来自不同扩增方法、不同测序平台、不同物种的单细胞转录组数据集),从而为复杂的生物过程提供新的理解。
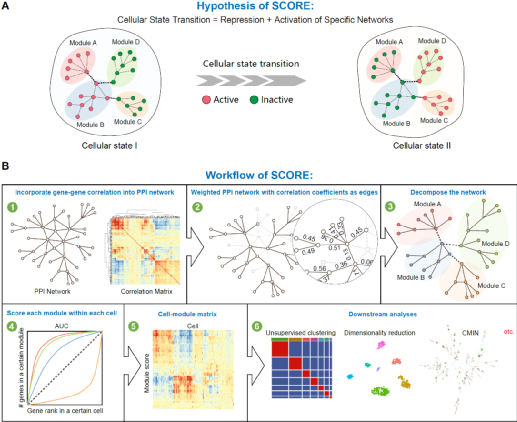
图一:SCORE 的工作流程。(A) SCORE 假设细胞状态的转变与分子网络功能模块的激活/抑制有关,这可以从单细胞转录组数据中推断出来。(B)SCORE将来自公共数据库的PPI分子网络和从单细胞数据集推断出的相关性分子网络相结合并修剪,从而构建加权分子相互作用网络。然后基于随机游走算法将该加权网络分解为多个功能模块,并利用 AUCell 计算每个细胞内的每个功能模块的激活分数。基于获得的细胞X功能模块激活分数的表达矩阵进行下游分析,从而对细胞进行聚类和可视化,并为每个细胞状态构建特征分子相互作用网络。
广州实验室研究员董骥博士、加州大学欧文分校周沛劼博士、北京大学吴艺翀博士以及陈依东博士为该论文的并列第一作者。北京未来基因诊断高精尖创新中心、北京大学生物医学前沿创新中心汤富酬教授和北京大学数学学院李铁军教授为该论文的共同通讯作者。该研究项目得到了国家自然科学基金委、北京市科技委和北京未来基因诊断高精尖创新中心的支持。

