
来源:ACS美国化学会
英文原题:DSDP: A Blind Docking Strategy Accelerated by GPUs
通讯作者: 高毅勤,北京大学
作者:YuPeng Huang, Hong Zhang, Siyuan Jiang, Dajiong Yue, Xiaohan Lin, Jun Zhang, Qinyi Gao*
背景介绍
分子对接是药物开发过程中发现候选分子的关键步骤,由确定位点、药物构象采样、打分、排序等步骤组成。在虚拟筛选任务中,一般通过靶蛋白和某些配体的共晶结构来确定结合位点的位置。然而,随着蛋白质三维结构预测方法的发展,越来越多不包含配体信息的蛋白质结构被预测出来。因此,在没有已知结合位点的情况下,仅基于蛋白质结构进行高精度的分子对接是非常必要的。
文章亮点
2023年6月29日,北京大学高毅勤教授团队在化学信息学和人工智能研究领域的国际权威学术期刊 Journal of Chemical Information and Modeling 上发表了题为“DSDP: A Blind Docking Strategy Accelerated by GPUs”的研究论文,该方法结合了机器学习与传统算法的优势,实现了 GPU 加速的高性能盲对接方法。与 AutoDock Vina 和 DiffDock 等分子对接程序相比,该方法在盲对接的速度与精度上均有显著提升。在不同测试集中,其成功率在高于 DiffDock 的前提下,速度提升70倍左右。
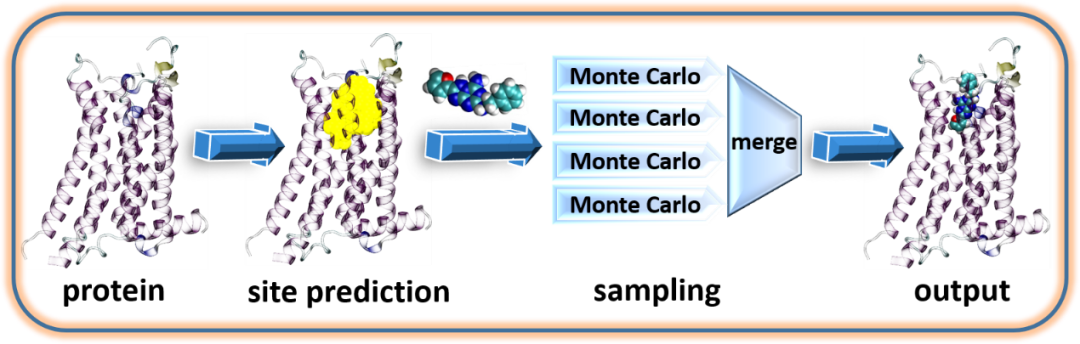
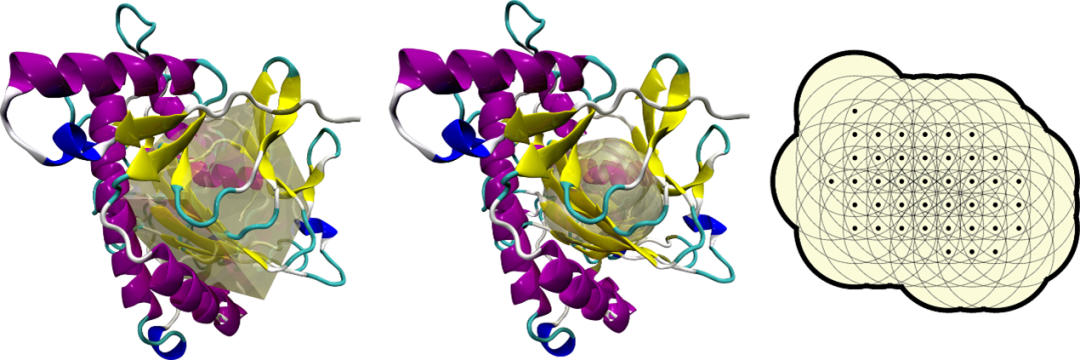
传统的分子对接程序在速度方面还有很大的提升空间,而机器学习虽然一定程度上解决了速度问题,但是受到蛋白质-小分子多构象数据集不足的限制,其对接精度还有待提升。为了将传统算法与机器学习的优势结合起来,该团队开发了 Deep Site and Docking Pose (DSDP) 方法,该方法主要包含两部分,一是基于机器学习的蛋白质结合位点预测,二是基于传统方法的构象采样及打分。前者使用了三维卷积神经网络,采用了18个维度的特征输入。后者使用了 Vina 的打分函数,采样部分针对 Vina 的思路进行了多处优化。位点预测的输出以散点的形式输入到采样过程中,提供了一个蛋白质口袋形状的采样空间,与传统的方盒子相比,减少了采样空间的浪费,提升了对接速度与精度。
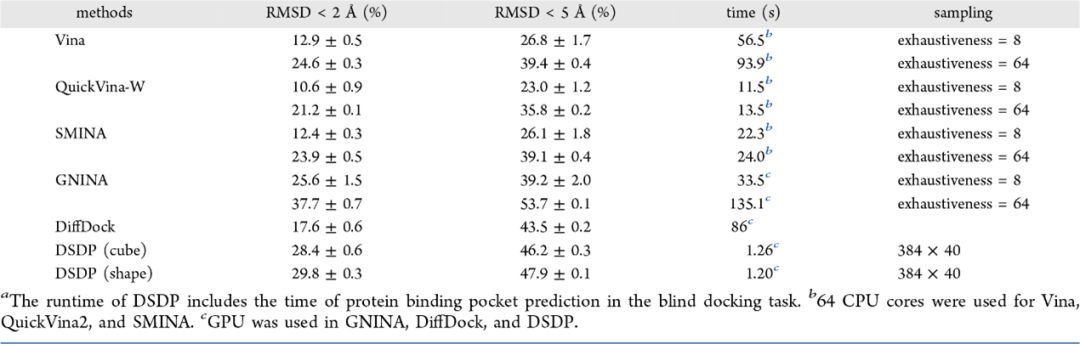
为了评估 DSDP 的性能,高毅勤教授团队提供了一个无偏数据集,包含995对蛋白-小分子复合物用于后续的性能测试。系统地比较 DSDP 与 AutoDock Vina、QuickVina、SMINA、GNINA、DiffDock 对接程序在盲对接任务中的表现。结果表明,在该测试集下除了64副本的 GNINA 以外,DSDP 的精度高于其他算法,每个体系平均速度为1.2 s是其他算法速度的几十甚至上百倍。GNINA 的高精度受益于机器学习的再打分过程,由此可见,优化打分函数是进一步提升对接精度的关键。此外,该工作还使用了 DiffDock 测试集以及 DUD-E 测试集,均表现出明显高于其他方法的综合性能。
DSDP 不仅可以执行盲对接任务,还可以完成虚拟筛选和再对接,该方法的开发极大地加速了虚拟筛选过程。在没有任何小分子结合位点信息的情况下,依然能实现高性能对接,这从一定程度上解决了未知靶点药物开发的难题。该程序已在github上开源:https://github.com/PKUGaoGroup/DSDP.git。

